 Introduction
IntroductionBackground
First described in 1922, Stevens-Johnson syndrome (SJS) is an immune-complex–mediated hypersensitivity complex that is a severe expression of erythema multiforme. It is known by some as erythema multiforme major, but disagreement exists in the literature. Most authors and experts consider SJS and toxic epidermal necrolysis (TEN) different manifestations of the same disease. For that reason, many refer to the entity as SJS/TEN. SJS typically involves the skin and the mucous membranes. While minor presentations may occur, significant involvement of oral, nasal, eye, vaginal, urethral, GI, and lower respiratory tract mucous membranes may develop in the course of the illness. GI and respiratory involvement may progress to necrosis. SJS is a serious systemic disorder with the potential for severe morbidity and even death. Missed diagnosis is common.
Although several classification schemes have been reported, the simplest breaks the disease down as follows:
- SJS - A "minor form of TEN," with less than 10% body surface area (BSA) detachment
- Overlapping SJS/TEN - Detachment of 10-30% BSA
- TEN - Detachment of more than 30% BSA
Pathophysiology
SJS is an immune-complex–mediated hypersensitivity disorder that may be caused by many drugs, viral infections, and malignancies. Cocaine recently has been added to the list of drugs capable of producing the syndrome. In up to half of cases, no specific etiology has been identified.
Pathologically, cell death results causing separation of the epidermis from the dermis. The death receptor, Fas, and its ligand, FasL, have been linked to the process. Some have also linked inflammatory cytokines to the pathogenesis.
Frequency
United States
Cases tend to have a propensity for the early spring and winter.
International
SJS occurs with a worldwide distribution similar in etiology and occurrence to that in the United States.
Mortality/Morbidity
- Mortality is determined primarily by the extent of skin sloughing. When BSA sloughing is less than 10%, the mortality rate is approximately 1-5%. However, when more than 30% BSA sloughing is present, the mortality rate is between 25% and 35%. See SCORTEN for a more complete discussion of severity of illness and mortality.
- Lesions may continue to erupt in crops for as long as 2-3 weeks. Mucosal pseudomembrane formation may lead to mucosal scarring and loss of function of the involved organ system. Esophageal strictures may occur when extensive involvement of the esophagus exists. Mucosal shedding in the tracheobronchial tree may lead to respiratory failure.
- Ocular sequelae may include corneal ulceration and anterior uveitis. Blindness may develop secondary to severe keratitis or panophthalmitis in 3-10% of patients. Vaginal stenosis and penile scarring have been reported. Renal complications are rare.
Race
A Caucasian predominance has been reported.
Sex
The male-to-female ratio is 2:1.
Age
Most patients are in the second to fourth decade of their lives; however, cases have been reported in children as young as 3 months.
Clinical
History
- Typically, the disease process begins with a nonspecific upper respiratory tract infection.
- This usually is part of a 1- to 14-day prodrome during which fever, sore throat, chills, headache, and malaise may be present.
- Vomiting and diarrhea are occasionally noted as part of the prodrome.
- Mucocutaneous lesions develop abruptly. Clusters of outbreaks last from 2-4 weeks. The lesions are typically nonpruritic.
- A history of fever or localized worsening should suggest a superimposed infection; however, fever has been reported to occur in up to 85% of cases.
- Involvement of oral and/or mucous membranes may be severe enough that patients may not be able to eat or drink.
- Patients with genitourinary involvement may complain of dysuria or an inability to void.
- A history of a previous outbreak of Stevens-Johnson syndrome (SJS) or of erythema multiforme may be elicited. Recurrences may occur if the responsible agent is not eliminated or if the patient is reexposed.
- Typical symptoms are as follows:
- Cough productive of a thick purulent sputum
- Headache
- Malaise
- Arthralgia
Physical
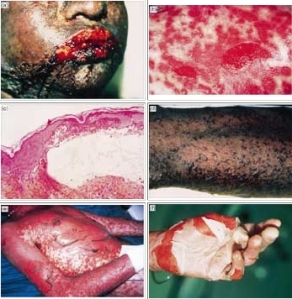
- The rash can begin as macules that develop into papules, vesicles, bullae, urticarial plaques, or confluent erythema.
- The center of these lesions may be vesicular, purpuric, or necrotic.
- The typical lesion has the appearance of a target. The target is considered pathognomonic. However, in contrast to the typical erythema multiforme lesions, these lesions have only two zones of color. The core may be vesicular, purpuric, or necrotic; that zone is surrounded by macular erythema. Some have called these targetoid lesions.
- Lesions may become bullous and later rupture, leaving denuded skin. The skin becomes susceptible to secondary infection.
- Urticarial lesions typically are not pruritic.
- Infection may be responsible for the scarring associated with morbidity.
- Although lesions may occur anywhere, the palms, soles, dorsum of the hands, and extensor surfaces are most commonly affected.
- The rash may be confined to any one area of the body, most often the trunk.
- Mucosal involvement may include erythema, edema, sloughing, blistering, ulceration, and necrosis.
- Although some have suggested the possibility of SJS without skin lesions, most believe that mucosal lesions alone are not enough to establish the diagnosis.
- The following signs may be noted on examination:
- Fever
- Orthostasis
- Tachycardia
- Hypotension
- Altered level of consciousness
- Epistaxis
- Conjunctivitis
- Corneal ulcerations
- Erosive vulvovaginitis or balanitis
- Seizures, coma
Causes
- Drugs and malignancies are most often implicated as the etiology in adults and elderly persons.
- Pediatric cases are related more often to infections than to malignancy or a reaction to a drug.
- A medication such as sulfa, phenytoin, or penicillin had previously been prescribed to more than two thirds of all patients with SJS. The anticonvulsant oxcarbazepine (Trileptal) has also been implicated. Hallgren et al reported ciprofloxacin-induced SJS in young patients in Sweden and commented on several others. Metry et al reported SJS in 2 HIV patients treated with nevirapine and mentioned one other in the literature. Metry et al speculated that the problem may extend to other non-nucleoside reverse transcriptase inhibitors. Indinavir has been mentioned. In 2007, the FDA issued a warning that SJS/TEN had occurred in patients taking modafinil (Provigil).
- More than half of the patients with SJS report a recent upper respiratory tract infection.
- The 4 etiologic categories are (1) infectious, (2) drug-induced, (3) malignancy-related, and (4) idiopathic.
- Viral diseases that have been reported include herpes simplex virus (HSV), AIDS, coxsackie viral infections, influenza, hepatitis, mumps, mycoplasmal infection, lymphogranuloma venereum (LGV), rickettsial infections, and variola.
- Bacterial etiologies include group A beta streptococci, diphtheria, Brucellosis,Mycoplasma pneumoniae, tularemia, and typhoid. mycobacteria,
- Coccidioidomycosis, dermatophytosis, and histoplasmosis are the fungal possibilities.
- Malaria and trichomoniasis have been reported as protozoal causes.
- In children, Epstein-Barr virus and enteroviruses have been identified.
- Drug etiologies include penicillins and sulfa antibiotics. Anticonvulsants including phenytoin, carbamazepine, valproic acid, lamotrigine, and barbiturates have been implicated. Mockenhapupt et al stressed that most anticonvulsant-induced SJS occurs in the first 60 days of use. In late 2002, the US Food and Drug Administration (FDA) and the manufacturer Pharmacia noted that SJS had been reported in patients taking the cyclooxygenase-2 (COX-2) inhibitor valdecoxib. In 2007, the US FDA reported SJS/TEN in patients taking modafinil (Provigil).
- Various carcinomas and lymphomas have been associated.
- SJS is idiopathic in 25-50% of cases.
 Introduction
Introduction
No comments:
Post a Comment